

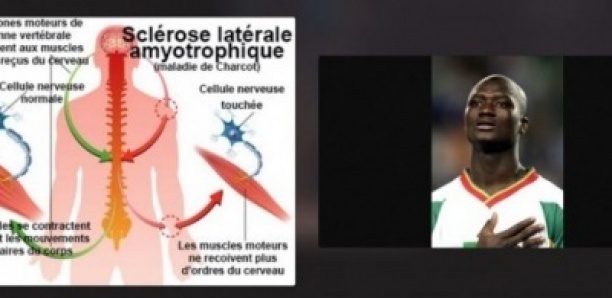
La Sclérose latérale amyotrophique (SLA) ou maladie de Charcot est liée à une dégénérescence des neurones moteurs.
La SLA est une maladie neurologique progressive touchant sélectivement les systèmes moteurs. La perte de motricité est la conséquence d’une dégénérescence, c’est-à-dire d’une mort cellulaire, des motoneurones, les cellules nerveuses (neurones) qui commandent les muscles volontaires.
L’atteinte concerne deux types de motoneurones qui sont situés à des niveaux différents du système nerveux : les motoneurones « centraux » et les motoneurones « périphériques » :
Les motoneurones centraux sont localisés dans le cerveau au niveau d’une région spécialisée dans la motricité appelée le cortex moteur : ils reçoivent l’ordre d’exécution du mouvement et le transmettent au tronc cérébral et à la moelle épinière.
Les motoneurones périphériques sont situés dans le tronc cérébral et la moelle épinière : ils sont directement connectés avec les muscles à qui ils transmettent l’ordre d’effectuer le mouvement.
Que signifie le terme Sclérose Latérale Amyotrophique (ou Maladie de Charcot) ?
L’appellation de SLA a été choisie pour décrire des anomalies constatées au niveau microscopique (« sclérose » et « latérale ») ou visibles à l’œil nu (« amyotrophique ») :
SCLEROSE : la dégénérescence des motoneurones laisse place à un tissu qui apparaît cicatriciel et fibreux
LATERALE : il existe une atteinte des fibres qui proviennent du motoneurone central et qui cheminent dans la partie latérale de la moelle
AMYOTROPHIQUE : la dégénérescence des motoneurones entraîne une fonte des muscles.
Le terme de « maladie de Charcot », du nom du neurologue français de la Pitié-Salpêtrière qui a décrit la maladie à la fin du 19ème siècle, est de moins en moins utilisée. Il avait l’inconvénient d’être à l’origine de confusions avec d’autres maladies décrites par Charcot.
Les appellations de « maladie du motoneurone » ou de « maladie de la corne antérieure » se rapportent à un ensemble plus large de maladies au cours desquelles on observe une atteinte des motoneurones. La sclérose latérale amyotrophique est la plus fréquente des maladies du motoneurone mais il existe d’autres affections, par exemple l’amyotrophie spinale de l’adulte ou la sclérose latérale primitive, dont les caractéristiques sont différentes.
La fréquence de la SLA
L’incidence de la SLA est de 1,5 nouveau cas par an et pour 100 000 habitants, ce qui représente environ 1000 nouveaux cas par an en France. Elle touche les deux sexes avec une légère prédominance masculine (rapport de 1,5).
Elle se déclare dans la majorité des cas entre 40 et 70 ans, l’âge moyen étant de 60 ans, mais elle peut survenir à tout âge chez l’adulte.
Il s’agit d’une maladie ubiquitaire. Il existe trois foyers de forte incidence de SLA qui sont tous situés dans les îles du pacifique ouest pour lesquels le rôle d’une toxine dans l’alimentation est suspecté.
Une maladie le plus souvent sporadique
La maladie est généralement sporadique mais il existe de rares formes familiales
Dans la grande majorité des cas, la maladie est sporadique, c’est-à-dire qu’elle survient isolément en l’absence d’autre cas de SLA dans la famille. Les formes familiales, définies par l’identification d’au moins 2 cas dans une généalogie, ne représentent que 5 à 10 % des cas de SLA. Ces formes sont généralement liées à l’intervention de facteurs génétiques. Il faut toutefois savoir que, étant donné le caractère non exceptionnel de la SLA, la survenue de deux cas dans une même famille peut être liée au hasard et n’implique donc pas nécessairement le caractère génétique de la maladie.
Les mécanismes de la SLA
L’origine primaire de la SLA sporadique est inconnue.
On ne connaît pas la cause initiale des formes sporadiques de SLA. La maladie est probablement multifactorielle, faisant intervenir des facteurs environnementaux et probablement également des facteurs de susceptibilité génétique. Certains facteurs environnementaux ont été suspectés mais aucun n’a pu être confirmé à ce jour.
Le rôle favorisant d’une activité physique importante ou de traumatismes répétés a été soulevé mais n’est pas prouvé. Quelles que soient les hypothèses formulées à partir d’études réalisées sur de larges populations de patients, il faut souligner qu’aucune donnée actuelle ne permet de remonter à une cause environnementale dans un cas individuel.
Les formes génétiques de la maladie représentent un groupe hétérogène.
Certaines sont liées au dysfonctionnement d’un gène unique (« formes monogéniques »). C’est dans ces formes que les progrès de la génétique ont permis d’identifier certains gènes responsables. L’anomalie la plus fréquente est une mutation dans le gène de la SOD1 (superoxyde dismutase de type 1) qui n’est toutefois responsable que d’environ 15 % de l’ensemble des formes génétiques. D’autres formes correspondent à une hérédité complexe, le développement de la maladie nécessitant probablement la présence d’anomalies de plusieurs gènes (« formes multigéniques »).
Cette forte hétérogénéité explique que devant une forme familiale de SLA, il n’est possible d’identifier une anomalie génétique que dans une petite minorité des cas.
Les mécanismes biologiques de la SLA sont de mieux en mieux compris
La maladie résulte d’une cascade d’événements biologiques multiples conduisant à la mort des motoneurones. Beaucoup de progrès ont été réalisés dans la caractérisation des anomalies, notamment grâce à l’analyse de modèles animaux de la maladie.
Les mécanismes de dégénérescence du motoneurone sont interdépendants et ont probablement une importance relative qui varie en fonction des stades de la maladie. On pense que les anomalies des mitochondries, les organites cellulaires qui produisent l’énergie nécessaire au métabolisme des motoneurones, jouent un rôle central. L’ensemble de ces mécanismes conditionne le motoneurone à entrer dans une voie finale de mort cellulaire où interviennent des phénomènes d’apoptose (ou « mort cellulaire programmée »).
Quelles sont les manifestations de la SLA ?
Les signes de la SLA découlent de l’atteinte sélective des motoneurones commandant les muscles volontaires (par opposition aux muscles qui ne peuvent être contrôlés par la volonté, comme le muscle cardiaque, qui sont respectés).
Selon le site où débute l’atteinte des motoneurones périphériques, on distingue deux formes de sclérose latérale amyotrophique :
la forme à début « spinal », liée à l’atteinte initiale des motoneurones de la moelle épinière, entraînant des troubles de la motricité des membres supérieurs ou inférieurs,
la forme à début « bulbaire », liée à l’atteinte initiale des motoneurones du tronc cérébral, provoquant des troubles de la parole et de la déglutition. Cette forme est plus fréquente chez la femme et débute généralement à un âge plus tardif.
Une caractéristique essentielle de la SLA est qu’en dehors de la motricité, elle respecte les autres fonctions du système nerveux. Les fonctions intellectuelles sont conservées tout le long de la maladie.
La SLA n’affecte également en rien les cinq sens, soit la vision, l’ouïe, le goût, l’odorat et le toucher. Elle n’affecte pas les muscles de l’œil, du cœur, de la vessie, de l’intestin et des organes sexuels. D’autres symptômes peuvent toutefois s’ajouter aux troubles moteurs : constipation, amaigrissement, douleurs, œdèmes et troubles vasomoteurs, troubles du sommeil et troubles respiratoires.
Un diagnostic basé sur l’examen neurologique et l’électromyogramme
Le diagnostic de SLA peut être difficile, notamment dans les formes de début où les signes sont incomplets. Il n’existe en effet pas de » marqueur » biologique de la maladie, c’est-à-dire d’anomalie sanguine caractéristique de la maladie. Le diagnostic repose sur l’expertise du neurologue qui va rechercher à l’examen des signes d’atteinte du motoneurone central (comme la raideur des muscles, l’exagération de certains réflexes ou la présence d’un signe de Babinski) ou périphérique (comme l’amyotrophie, les crampes, l’abolition de réflexes ou les fasciculations qui sont des contractions involontaires de fibres musculaires visibles à la surface du muscle).
Cet examen est complété par un électromyogramme qui recherche des anomalies électriques de souffrance du motoneurone périphérique et précise leur extension.
Les autres examens sont réalisés au cas par cas et ont pour but d’éliminer une autre cause pouvant éventuellement expliquer les troubles moteurs. Une IRM du cerveau et/ou de la moelle est souvent réalisée. Une ponction lombaire ou une biopsie musculaire ne sont en revanche nécessaires que dans une minorité de cas.
Ce bilan permet dans la majorité des cas d’établir le diagnostic avec certitude. Toutefois, il peut arriver qu’en l’absence de signes suffisamment caractéristiques il soit seulement possible de poser un diagnostic de probabilité. C’est alors seulement le suivi du patient, avec la répétition des examens cliniques et au besoin électro-myographiques, qui permettra de confirmer ou d’infirmer le diagnostic initial.
Une évolution imprévisible
La sclérose latérale amyotrophique est une maladie handicapante qui évolue progressivement.
Le degré d’évolution de la maladie peut être évalué par des tests cliniques simples et par un bilan respiratoire régulier afin d’évaluer une atteinte éventuelle des muscles respiratoires.
Le profil évolutif de la maladie varie en effet d’un patient à l’autre et également chez un même patient au cours du temps. La maladie peut se stabiliser spontanément (environ 15 % des patients) à un stade de handicap variable.
Il est a priori impossible de prédire l’évolution de cette maladie chez un patient donné.
Le traitement neuroprotecteur ralentit l’évolution de la maladie
Il n’existe pas de traitement permettant de guérir, ni même de stopper l’évolution de la SLA. Un médicament, le riluzole permet de ralentir l’évolution de la maladie, ce qui a été confirmé par de nombreuses études. Il agit notamment sur le métabolisme du glutamate qui est perturbé au cours de la SLA.
Il s’agit d’un médicament à prendre par voie orale qui entraîne peu d’effets secondaires (une fatigue et des troubles intestinaux transitoires sont parfois observées en début de traitement). Le traitement nécessite toutefois une surveillance par des prises de sang régulières (une élévation des enzymes du foie est possible).
La vitamine E peut être prescrite en raison de ses propriétés anti-oxydantes bien que les études réalisées chez l’homme n’aient pas démontré d’effets significatifs sur l’évolution de la maladie. De nouveaux traitements sont régulièrement testés dans la SLA.
Leur évaluation nécessite d’être faite dans le cadre d’essais thérapeutiques rigoureux sous une surveillance étroite des effets secondaires possibles.
→ A LIRE AUSSI : Décès de Pape Bouba Diop: sur chaise roulante depuis deux ans, il devait être au Sénégal cette semaine
→ A LIRE AUSSI : Laye Diaw fait des révélations surprenantes sur le décès de Pape Bouba Diop (audio)
→ A LIRE AUSSI : Les Lions du Sénégal rendent hommage à Papa Bouba Diop: “La Tanière est effondrée”